2 — La fluorescence
2.1 Photoluminescence
La photoluminescence désigne l'émission de lumière par une molécule après absorption d'un photon. Elle regroupe deux phénomènes distincts selon la nature de l'état excité impliqué :
En microscopie de fluorescence, on exploite exclusivement la fluorescence pour sa rapidité, sa spécificité et sa reproductibilité.
2.2 Diagramme de Jablonski
Le diagramme de Jablonski représente les niveaux d'énergie électroniques et vibrationnels d'un fluorophore. Cliquez sur chaque flèche pour en savoir plus.
2.3 Décalage de Stokes
Après l’absorption d’un photon d’excitation, la molécule excitée perd une partie de son énergie par relaxation vibrationnelle, sous forme de chaleur, avant l’émission lumineuse. Le photon émis (hν') est donc moins énergétique que le photon absorbé (hν), ce qui correspond à une longueur d’onde plus grande :
Cette différence de longueur d’onde, appelée décalage de Stokes (Δλ), permet de séparer spectralement l’excitation de l’émission,
un point essentiel pour filtrer le signal fluorescent en microscopie.
Simulation — Excitation et collecte du signal de la GFP
Cette simulation illustre l’impact du choix de la longueur d’onde d’excitation et du filtre d’émission sur l’intensité du signal fluorescent collecté. Déplacez le laser et le filtre pour voir leur impact sur le signal fluorescent collecté.
Cellule
2.4 Caractéristiques d'un fluorophore
| Paramètre | Symbole | Définition | FITC vs Alexa647 |
|---|---|---|---|
| Coefficient d'absorption molaire | ε | Capacité à absorber les photons à une longueur d’onde donnée (L·mol⁻¹·cm⁻¹) | FITC : 73 000 / Alexa647 : 270 000 |
| Efficacité quantique | QY | Fraction des photons absorbés qui sont réémis (0–1) | FITC : 0,92 / Alexa647 : 0,33 |
| Brillance | ε × QY | Signal émis par molécule — indicateur direct de la détectabilité en microscopie | FITC : 67 000 / Alexa647 : 89 100 |
| Lifetime | τ | Temps à 1/e de l'intensité après excitation pulsée (ns) | FITC : ~4 ns / Alexa647 : ~1 ns |
| Photostabilité | — | Résistance au photoblanchiment sous illumination continue | Alexa Fluor >> FITC |
2.5 Types de fluorophores
Fluorophores organiques
Petites molécules synthétiques (Alexa Fluor, FITC, DAPI, Cy3, Cy5…). Grande variété spectrale, haute brillance. Nécessitent une fixation ou une conjugaison à un anticorps / ligand. Certains sont capables de traverser la membrane plasmique, d'autres ont besoin que cette dernière soit perméabilisée.

Protéines fluorescentes
Majoritairement dévirées de la GFP, elles peuvent être integrées au génome par ingénérie moléculaire, et donc être exprimées dans des organismes vivants. Elles peuvent être fusionnées à une protéine d'interet pour la localiser ou suivre son déplacement.
.jpg)
Quantum dots
Nanocristaux semi-conducteurs (CdSe, InP…). Spectre d'émission très étroit accordable par la taille du nanocristal. Spectre d'excitation large. Excellente photostabilité — idéaux pour le multiplexage et le suivi longue durée.
2.6 Séparation excitation / émission dans un microscope à fluorescence
En microscopie à fluorescence, le principal défi expérimental consiste à séparer la faible lumière émise par le fluorophore de la lumière d’excitation, beaucoup plus intense. Cette séparation est rendue possible grâce au décalage de Stokes et à une combinaison de filtres spectraux et d’un miroir dichroïque.
- Filtre d’excitation — sélectionne une bande étroite de longueurs d’onde correspondant au spectre d’absorption du fluorophore (ex. 488 nm pour la GFP).
- Miroir dichroïque — réfléchit la lumière d’excitation vers l’échantillon et transmet la lumière d’émission vers le détecteur.
- Filtre d’émission — bloque la lumière d’excitation résiduelle et ne laisse passer qu’une bande spectrale centrée sur l’émission du fluorophore (ex. 507 ± 15 nm pour la GFP).
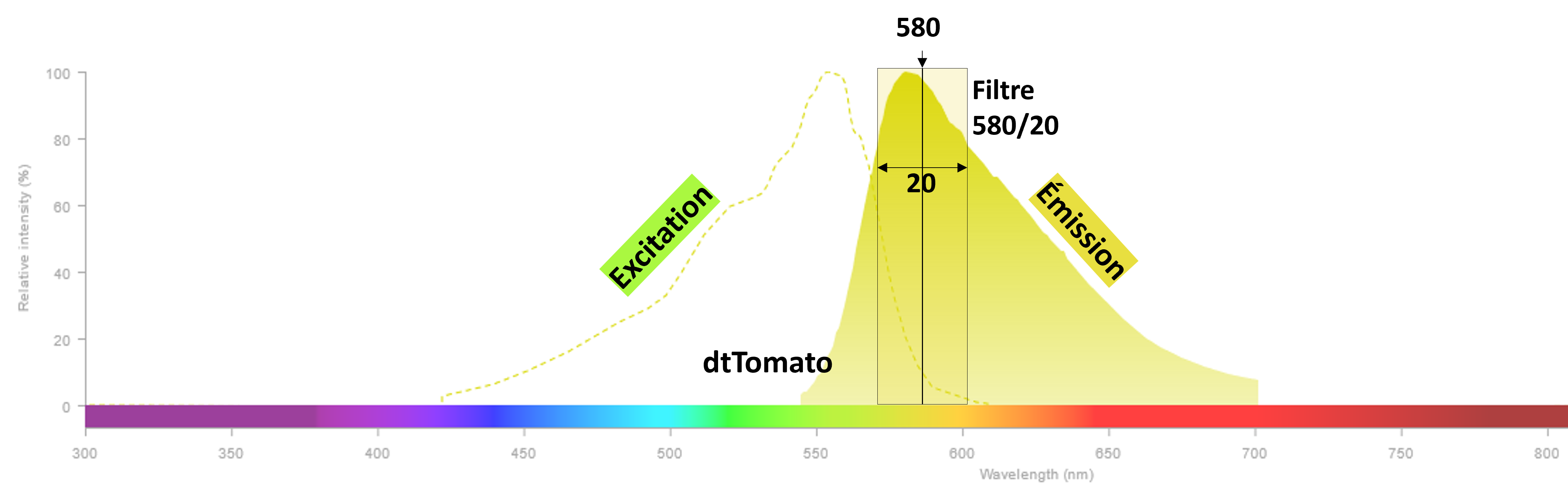
Les filtres d'émission/détection sont caractérisés par deux valeurs : la première correspond à la longueur d'onde au
centre du filtre, et la seconde à la large du filtre. Ainsi, un filtre 580/20 est centré sur 580nm et a une largeur
totale de 20nm. Il laisse donc passer la lumière de 570 à 590nm.
Cette notation est universelle.
2.7 Photoblanchiment
Le photoblanchiment (photobleaching) est la perte irréversible de fluorescence par destruction du fluorophore. Une excitation trop forte, une exposition prolongée ou répétée, ou des conditions environnementales défavorables (pH, température, présence d'oxygène) peuvent altérer irrémédiablement la structure du fluorophore et supprimer toute émission.
- Réduire la puissance laser au minimum nécessaire
- Limiter le temps d'exposition et le nombre d'acquisitions
- Utiliser des agents antifade (Mowiol + DABCO, ProLong Gold, Trolox)
2.8 Autofluorescence
Certaines molécules biologiques possèdent une fluorescence intrinsèque : elles émettent naturellement de la lumière lorsqu'elles sont excitées par un laser. C'est l'autofluorescence. Les principales sources d'autofluorescence en biologie cellulaire sont le NADH, le FAD et la kératine dans les tissus. D'autres molécules peuvent contribuer selon le type d'échantillon. Ceci pose un problème, car cette autofluorescence génère un signal en plus de celui de nos fluorophores, ce qui contribue à diminuer le ratio signal/bruit : c'est le bruit de fond. On l'observe davantage dans les courtes longueurs d'ondes et de moins en moins lorsqu'on regarde vers le rouge. Pour y remédier, on peut sélectionner des fluorophores émettant dans le rouge, utiliser des contrôles négatifs pour caractériser le bruit de fond, et limiter l'excitation.