6 — Conception et acquisition
6.1 — Design expérimental
Toute experience de microscopie confocale commence par le choix des fluorophores. En effet, il faut que ces derniers soient adaptés à l'équipement disponible. Il n'est pas rare de trouver des microscopes confocaux équipés au moins des quatre lasers suivants : 405 nm (violet), 488 nm (bleu), 561 nm (vert) et 647 nm (rouge). Cette configuration "4 couleurs" couvre un grand nombre des fluorophores disponibles sur le marché. Évidemment, certains systèmes peuvent avoir moins de lasers, d'autres davantage.
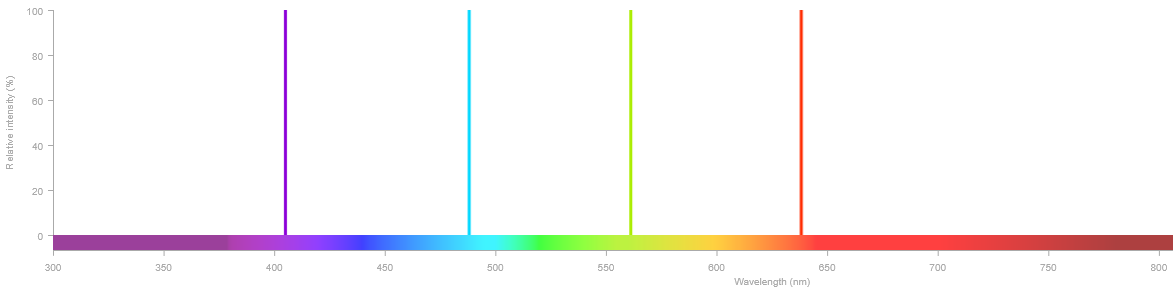
Il existe également des sources laser à spectre continu, comme le laser à lumière blanche (WLL), capables d'émettre à n'importe quelle longueur d'onde dans une plage donnée (typiquement 440–790 nm). Cela permet d'exciter précisément au pic d'absorption de n'importe quel fluorophore, réduisant la puissance nécessaire et donc le photoblanchiment.
Pour chaque laser, un filtre d'émission (voir 2.6) sélectionne la bande spectrale détectée. Le choix du bon couple laser/filtre pour chaque fluorophore est donc essentiel avant de démarrer l'acquisition.
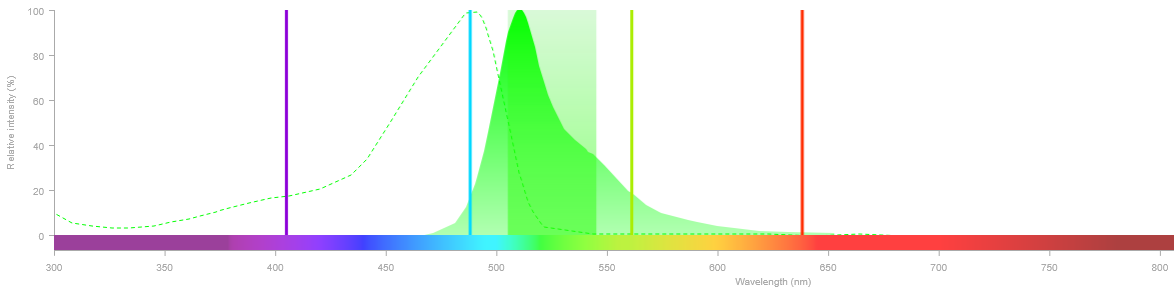
En pratique, plusieurs outils en ligne permettent de visualiser les spectres d'excitation et d'émission des fluorophores et de vérifier leur compatibilité avec votre équipement :
- Spectraviewer — Thermo Fisher
- Spectraviewer — Chroma
- FPbase — base de données des protéines fluorescentes
- Fluorophore Selection Guide — Thermo Fisher
- Le DAPI est un fluorophore couramment utilisé pour marquer l'ADN, et donc les noyaux des cellules. Son pic d'excitation est autour de 360nm, mais en microscopie confocale il est classique d'utiliser le laser violet (405nm) pour l'exciter. Bien que loin de la condition idéale, la brillance du DAPI est si élevée (voir 2.4) qu'il produit un signal très satisfaisant dans ces conditions.
- Ce cas particulier illustre un principe général : un fluorophore dont le pic d'excitation ne correspond pas exactement au laser disponible n'est pas nécessairement à exclure. Sa brillance peut compenser une excitation sous-optimale et rendre son utilisation tout à fait satisfaisante. Il vaut donc toujours la peine de vérifier ce paramètre avant d'écarter un fluorophore.
6.2 Crosstalk et acquisition séquentielle
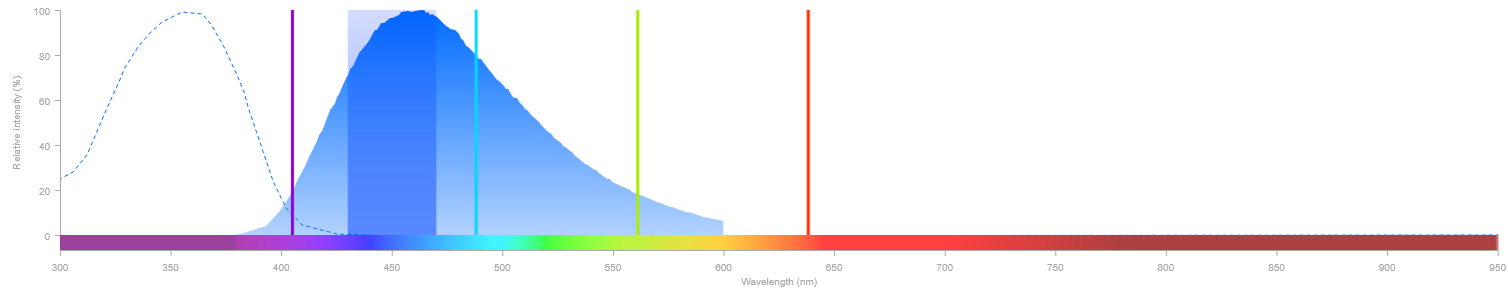
Lorsque plusieurs fluorophores sont utilisés simultanément dans une même acquisition, il peut exister un risque de contamination spectrale (crosstalk) entre eux. Cela entraîne alors une détection d'un fluorophore dans le canal d'un autre, ce qui fausse l'interprétation de l'image. Deux situations peuvent en être la cause :
- Excitation non spécifique — un laser excite partiellement un fluorophore pour lequel il n'est pas destiné.
- Débordement spectral (bleed-through) — l'émission d'un fluorophore déborde dans la fenêtre de détection d'un autre.
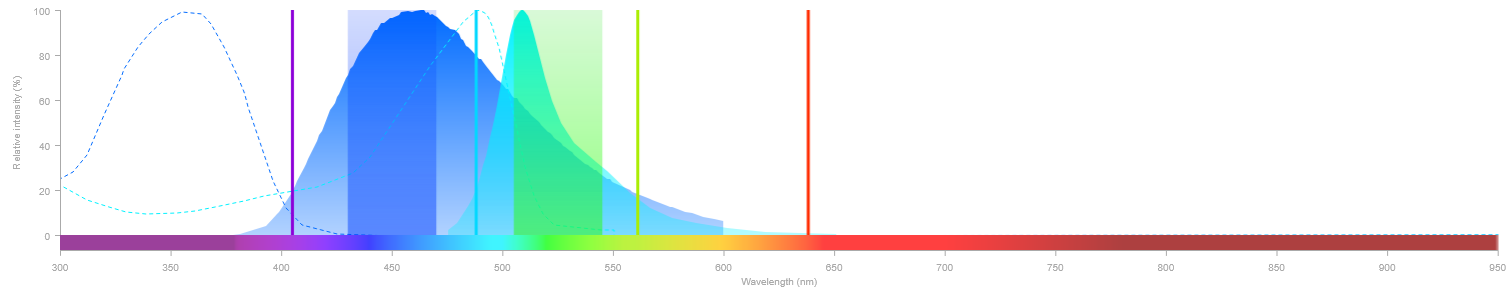
La solution est d'acquérir les canaux en série plutôt que simultanément : on active un seul laser à la fois et on collecte le signal correspondant avant de passer au suivant. Cela élimine toute excitation non spécifique et réduit drastiquement le bleed-through.
6.3 Profondeur de champ
La profondeur de champ correspond à l'épaisseur de l'échantillon qui apparaît nette simultanément dans l'image. Elle est inversement proportionnelle à l'ouverture numérique (NA) de l'objectif (voir 4.3).

© Gary Holpin, garyholpin.co.uk
À forte ouverture numérique (NA ≥ 1.2), la profondeur de champ est d'environ 0.8 µm, bien plus petite que l'épaisseur d'une cellule. Une seule image ne capture donc qu'une fine tranche de cette cellule.
La profondeur de champ souhaitée dépend avant tout de la question biologique. Une grande profondeur de champ (= faible NA, donc souvent faible grossissement) donne une vue globale de l'échantillon en une seule image, tout apparaissant net simultanément. Une faible profondeur de champ (= haute NA, donc souvent fort grossissement) permet de mettre en focus une structure précise à une profondeur donnée — le reste de l'échantillon apparaît flou mais contribue toujours au signal. En confocal, le pinhole va plus loin en rejetant physiquement ce signal hors-focus, permettant ainsi des coupes optiques nettes et la reconstruction 3D.
6.4 Z-stack et reconstruction 3D
En microscopie confocale, à pinhole fermé, une seule image ne capture qu'une fine tranche de l'échantillon. L'acquisition de plusieurs images les unes au-dessus des autres dans la profondeur Z, permet la reconstruction d'un volume complet. C'est ce qu'on appelle un Z-stack.
Comme pour l'échantillonnage en XY (voir 5.3), l'intervalle entre les coupes en Z doit respecter le critère de Nyquist. La résolution axiale étant environ 2 à 3 fois inférieure à la résolution latérale, l'intervalle Z optimal est typiquement de l'ordre de 200–300 nm pour un objectif dont la résolution XY est 70-100nm. La plupart des microscopes modernes calculent et suggèrent automatiquement cet intervalle.
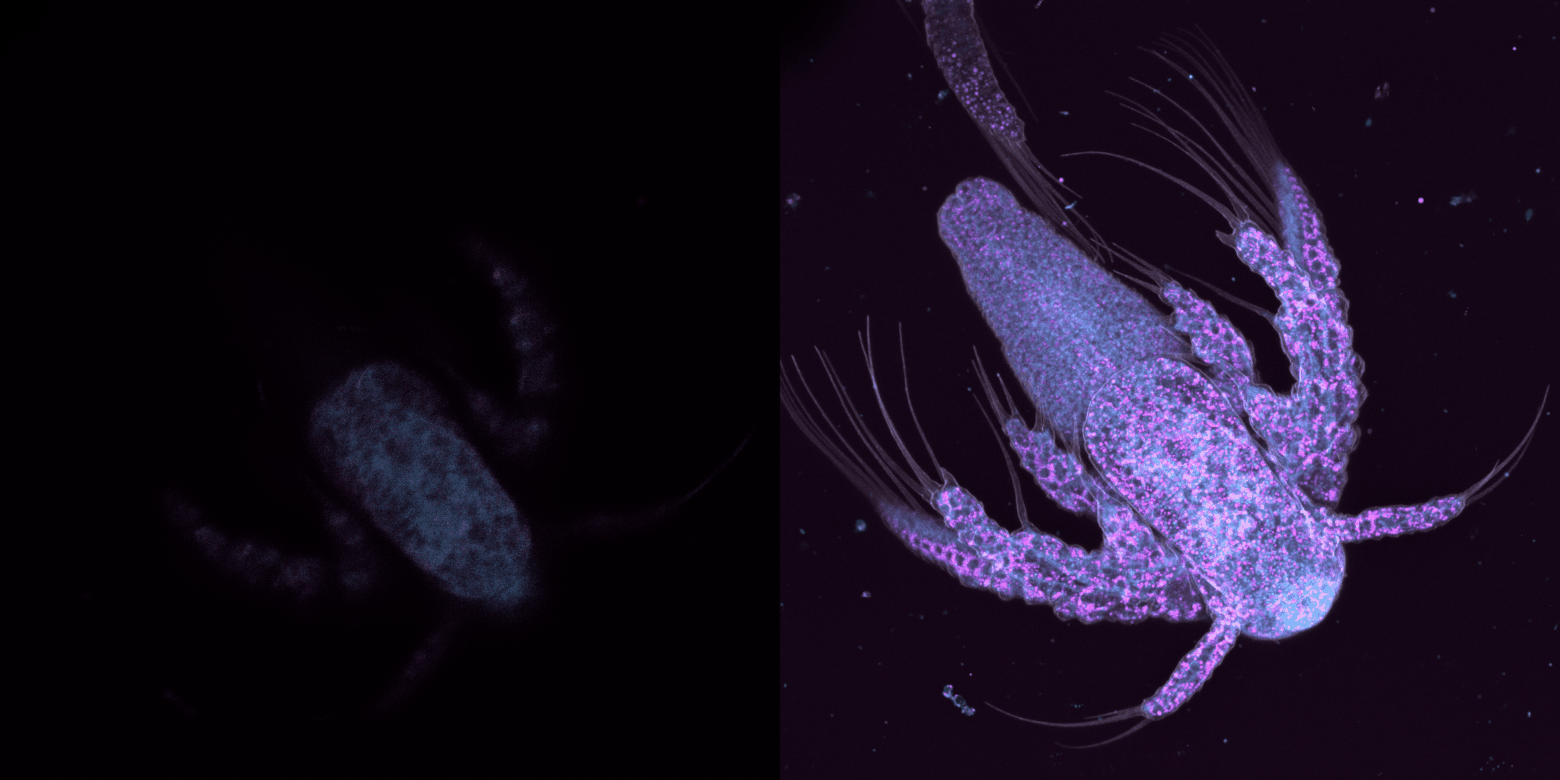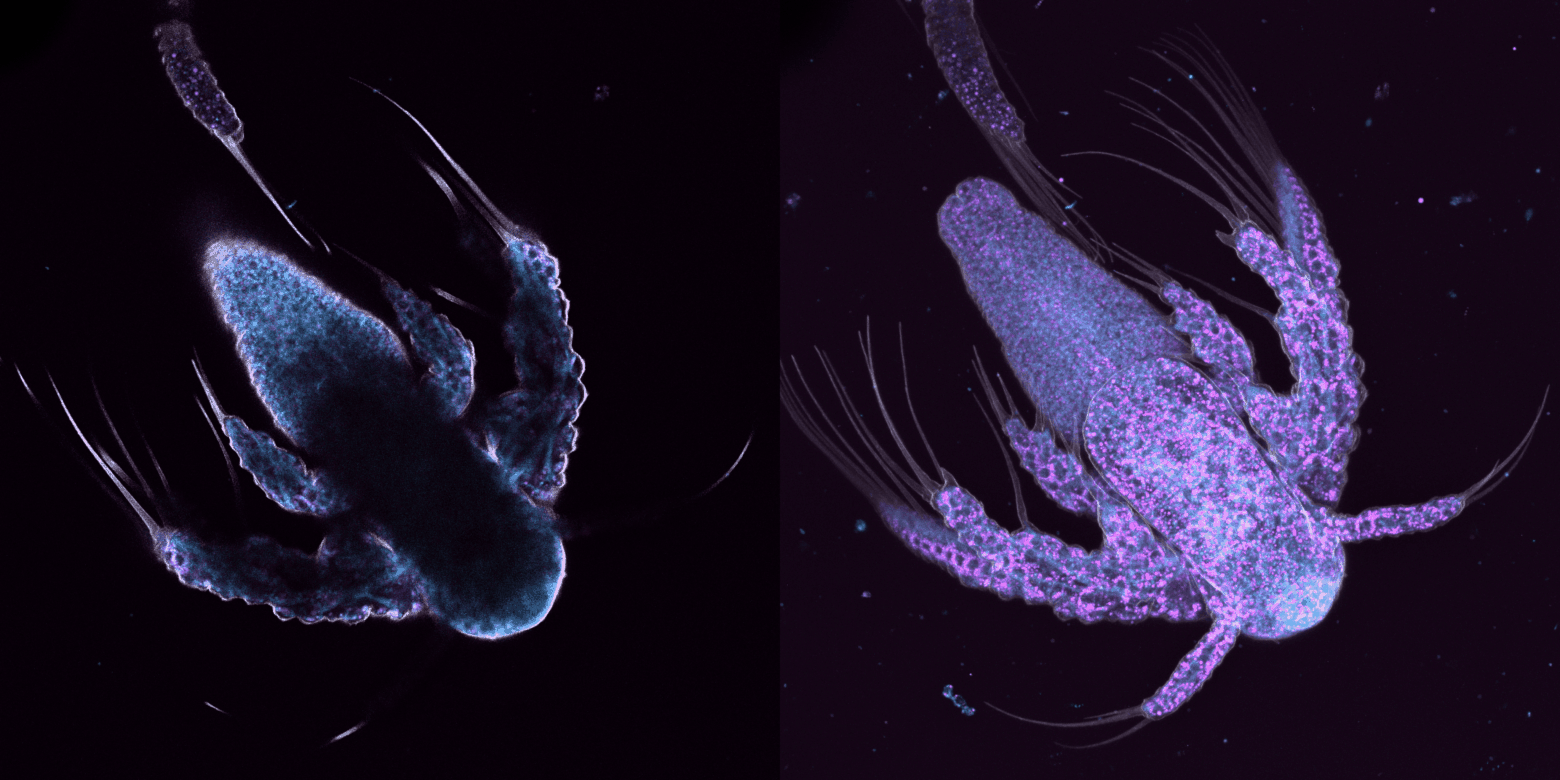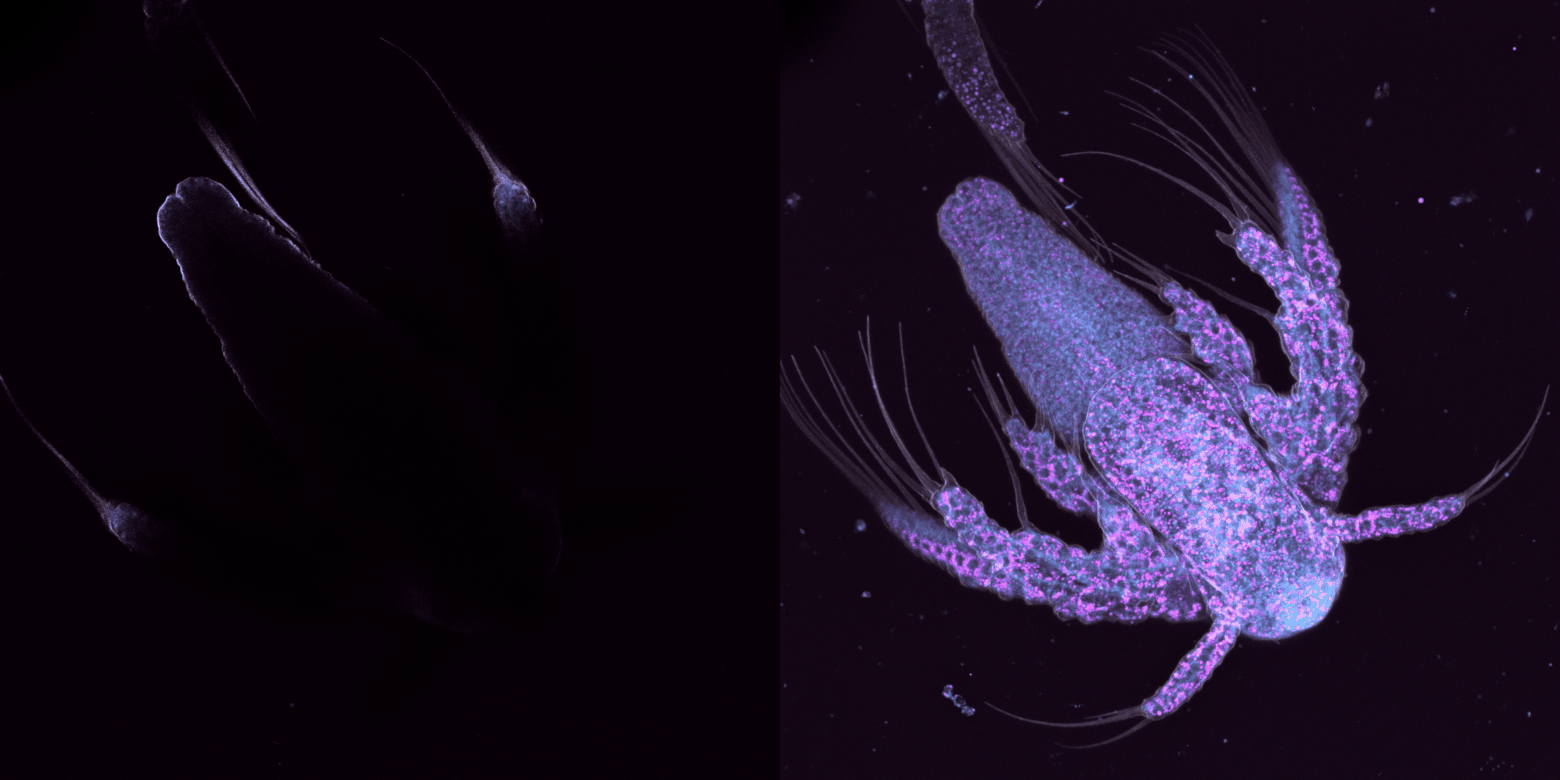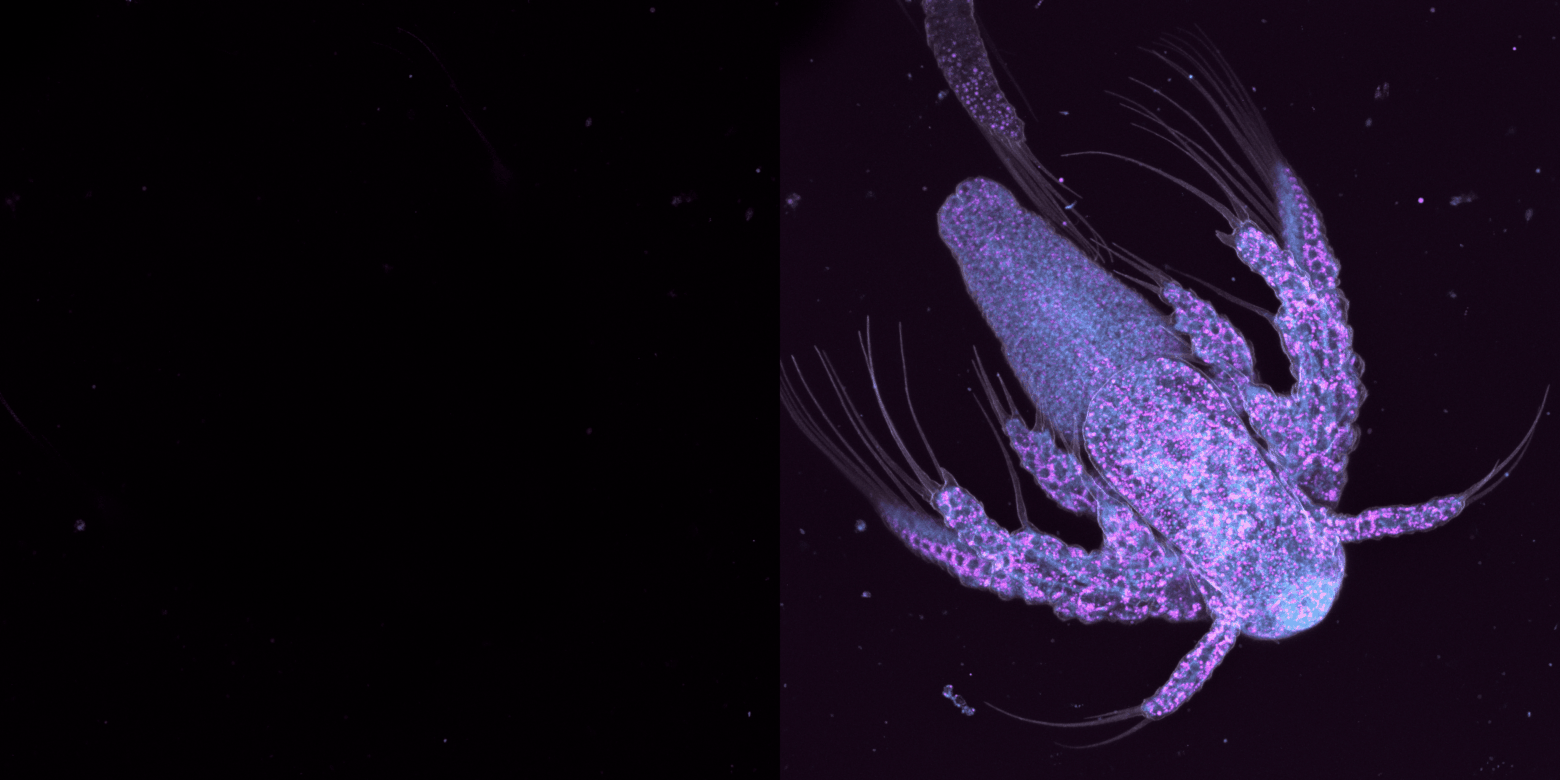
trop large, on perd du temps à capturer des images sans intérêt et on photoblanchit notre échantillon, trop étroit, on risque de rater de l'information.
6.5 Déconvolution
Même avec un confocal et un pinhole bien réglé, chaque point de l'échantillon reste un peu déformé par la PSF de l'objectif (voir 5.1). L'image acquise n'est donc pas une représentation parfaite de l'échantillon, mais ce que la PSF nous en montre.
Puisque la PSF est connue mathématiquement (voir 5.1), il est possible de calculer son inverse et de corriger numériquement ce flou : c'est la déconvolution. En pratique, elle permet d'améliorer la résolution latérale de ~200 nm à ~140 nm, d'augmenter le contraste et de réduire le bruit de fond.

La déconvolution présente cependant des limites : elle nécessite un Z-stack complet (pas applicable sur une image seule), un temps de calcul important (accéléré par GPU), et un logiciel dédié parfois coûteux. Elle peut également introduire des artefacts si mal paramétrée.